Qu’est-ce que le syndrome de Sturge-Weber ?
Le syndrome de Sturge-Weber est une maladie rare présente dès la naissance. Il s’agit d’une anomalie des vaisseaux sanguins qui peut toucher la peau, le cerveau et les yeux selon l’emplacement de l’atteinte.
Selon les personnes, le syndrome peut entraîner différents syndromes neurologiques ou oculaires qui nécessitent un suivi médical spécialisé.
Chaque personne atteinte est différente : certaines ont peu de symptômes, tandis que d’autres nécessitent un suivi plus important.
Le syndrome de Sturge-Weber est une maladie rare. On estime qu’il touche environ 1 naissance sur 20 000 à 50 000.
En raison de sa rareté, les familles peuvent parfois se sentir isolées. C’est pour cela que les associations et les centres spécialisés jouent un rôle important pour informer et accompagner les patients.
Le syndrome de Sturge-Weber est lié à une mutation génétique survenue très tôt pendant le développement du bébé.
Cette mutation concerne un gène appelé GNAQ ou GNA11.
Il est important de savoir que :
- Ce n’est pas une maladie héréditaire
- Les parents n’y sont pour rien
- La mutation apparaît de manière spontanée


Les signes cliniques
Les manifestations du syndrome peuvent varier d’une personne à l’autre. Elles peuvent concerner :
La peau
Angiome plan sur le visage
Le cerveau
Crises d’épilepsie
Faiblesse d’un coté du corps
Difficultés d’apprentissage chez certains enfants
Les yeux
Glaucome
Augmentation de la pression dans l’œil
Le syndrome de Sturge-Weber peut se présenter sous différentes formes selon les zones touchées.
Les médecins distinguent généralement trois types principaux d’atteinte, présentés ci-dessous :
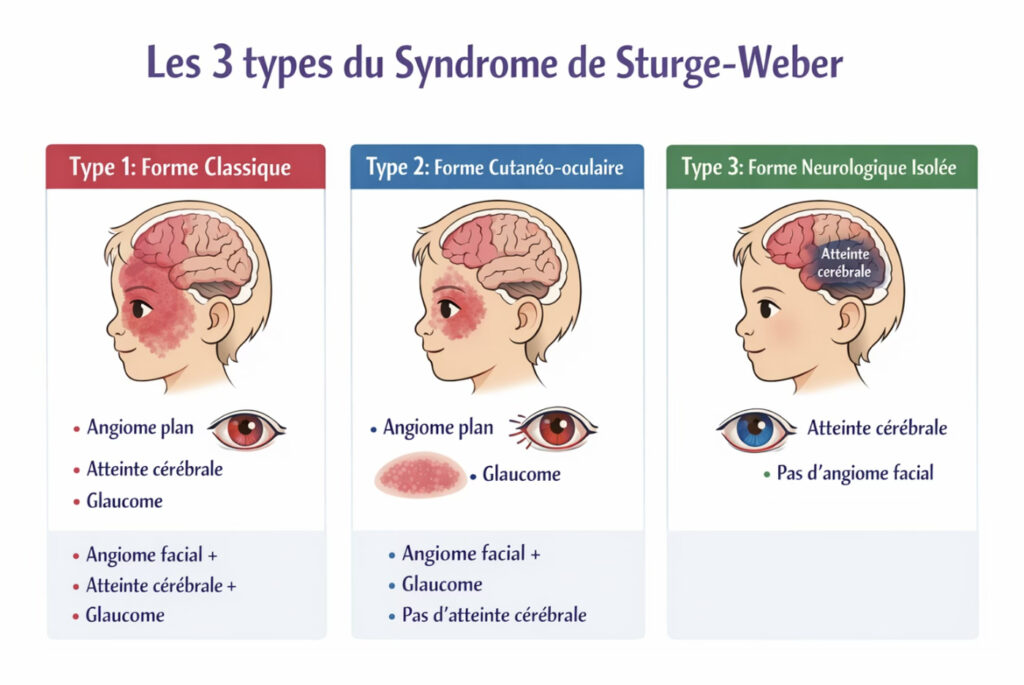
Chaque personne atteinte est unique. Toutes les manifestations ne sont pas forcément présentes et leur intensité peut varier d’un patient à l’autre.
Angiome plan
L’angiome plan, souvent appelé « tache de vin », est l’un des signes les plus visibles du syndrome de Sturge-Weber. Il s’agit d’une malformation des petits vaisseaux sanguins située dans la peau.
Cet angiome est présent dès la naissance et apparaît généralement sous la forme d’une tache rouge ou violacée sur le visage. Il se situe le plus souvent sur le front ou autour de l’œil, mais sa taille et sa localisation peuvent varier d’une personne à l’autre.
L’angiome plan n’est pas douloureux et ne présente pas de danger en lui-même. Cependant, lorsqu’il se trouve dans certaines zones du visage, notamment autour de l’œil ou du front, il peut être associé au syndrome de Sturge-Weber et nécessiter un suivi médical.
Pourquoi apparaît-il ?
L’anigome plan est lié à une anomalie dans le développement des vaisseaux sanguins. Dans le syndrome de Sturge-Weber, cette anomalie est due à une mutation génétique qui survient très tôt pendant le développement du bébé.
Le suivi et les traitements
Dans certains cas, un traitement par laser dermatologique peut être proposé pour éclaircir l’angiome.
Ce traitement est principalement esthétique, mais il peut aussi avoir un impact positif sur le bien-être et la confiance en soi.
Chaque situation est différente et les médecins adaptent la prise en charge en fonction de l’âge de l’enfant, de la localisation de l’angiome et des besoins de la famille.
Nous avons posé quelques questions au Docteur Christine Léauté-Labrèze de l’unité de dermatologie pédiatrique, Centre de référence des maladies rares de la peau – MAGEC Sud ; CHU de Bordeaux, au sujet du laser dermatologique :
En quoi consiste le laser dermatologique ?
Un laser (light Ampilification by Stimulated Emissions of Radiation) est une source de lumière émettant une seule longueur d’onde et dans une seule direction, ce qui permet d’obtenir une tâche lumineuse très petite et donc très intense. Les laser dits dermatologiques ont des longueurs d’onde particulières leur permettant de traverser la peau et atteindre leur cible. Pour traiter les angiomes plans, on utilise préférentiellement la longueur d’onde de 595nm lumière jaune/orangée), la cible est constituée par l’oxyhémoglobine (pigment rouge) des globules rouges (cellules qui circulent dans les vaisseaux et transportent l’oxygène dans l’organisme). Quand la lumière atteint sa cible, la chaleur est telle que le petit vaisseau situé dans la peau va être détruit. L’avantage est que la surface de la peau n’est pas endommagée et qu’il n’y a donc pas de risque de brûlures et donc de cicatrices. Par ailleurs comme on utilise de la lumière visible il n’y a pas de risque de cancers de la peau à long terme, contrairement à l’exposition aux rayons X ou le rayonnement ultraviolets (UVA et UVB).
A quel âge peut-on commencer, il y a-t-il un âge limite préconisé ?
Le traitement peut être commencé très jeune à quelques semaines de vie, l’avantage est que la surface de l’angiome à traiter est petite (seulement quelques impacts laser), néanmoins il faut tenir compte du fait que le traitement est douloureux et qu’il est fortement déconseillé de faire des procédures itératives douloureuses à un enfant (risque de retentissement émotionnel important). Personnellement, je ne suis pas rigide concernant l’âge pour commencer le laser, il faut en discuter et voir ce qui est le mieux pour l’enfant, par exemple si l’enfant fait de nombreuses crises convulsives il faut attendre que la situation soit plus stable pour ne pas lui faire courir de risque, on peut aussi attendre que l’enfant soit en âge de donner son avis pour être traité.
Est-ce qu’une séance est douloureuse ?
Oui le traitement est douloureux, on compare l’impact laser à un coup d’élastique tiré à bout portant sur la peau. Ce qui veut dire que chez un petit enfant, on peut faire quelques impacts avec une crème anesthésique, mais quand la surface est importante (imaginez recevoir une centaine de coups d’élastiques sur le visage) on peut avoir recours à une anesthésie générale (dans le cas d’un syndrome de Sturge-Weber on peut aussi profiter de l’anesthésie pour faire l’examen ophtalmologique et la prise de tension oculaire).
Il y a-t-il une sensation de brûlure après l’intervention au laser ?
Il y a une sensation de brûlure pendant 24 à 48 heures, mais avec une crème apaisante et du paracétamol, les suites sont habituellement simples.
Peut-on dire que l’efficacité du laser est systématique ?
On ne peut pas dire que l’efficacité est systématique. En effet, en cas d’angiome plan très marqué (rouge foncé ou violet), cela veut dire que les vaisseaux dans la peau sont assez gros et que le laser ne sera pas assez puissant pour les détruire (il y a trop de globules rouges), a contrario, si l’angiome est plutôt rose pâle les vaisseaux qui le constituent sont très fins et il n’y a pas beaucoup de globules rouges à l’intérieur et on n’a donc pas une bonne cible. Par ailleurs, il y a des régions du corps qui réagissent mieux que d’autres, au niveau du visage, les angiomes situés sur le front et les tempes pâlissent bien après une ou deux séances, alors que les joues ou le nez font souvent de la résistance et restent assez colorés malgré plusieurs passages de laser. On obtient également de bons résultats sur le tronc, mais il y a peu d’effets sur les extrémités, notamment les jambes, les mains et les pieds.
Combien de séances en moyennes faut-il faire ?
Pour un visage on compte en moyenne 4 à 6 séances pour obtenir un résultat significatif, mais si l’enfant n’a qu’une atteinte du front, 2 séances peuvent être suffisantes alors que pour une atteinte de la joue on fera peut-être plus de séances. Il faut évaluer le résultat après chaque séance et discuter de la suite à donner. Parfois le traitement laser ne donne pas le résultat escompté, dans ce cas il faut parfois faire une pause, certains enfants sont lassés et ont besoin de faire « un break », d’autres assument complètement leur différence, je leur dis que la porte reste ouverte et ils reviennent quand ils veulent (parfois c’est à l’adolescence).
Combien de temps met la peau pour cicatriser des impacts du laser ?
Avec le laser la surface de la peau n’est pas affectée, il n’y a pas besoin de pansements ou de soins compliqués. Comme on fait éclater les petits vaisseaux, il y a une hémorragie sous la peau qui donne cette couleur gris-violet (purpura) et qui va se résorber en 7 à 15 jours. On estime qu’après 3 mois on a le résultat final de la séance laser.
Est-ce normal que la zone traitée soit gonflé après séance ?
Oui, car on détruit les vaisseaux de la peau, cela créé de l’inflammation et il y a un afflux de cellules et de lymphe qui viennent en quelque sorte « faire du nettoyage » pour évacuer les cellules des vaisseaux qui ont été détruits. Le gonflement est particulièrement important quand on traite le visage (œdème des paupières).
Quel impact peut avoir le soleil sur la peau avant et après intervention ?
L’exposition au soleil entraine le bronzage, un pigment (la mélanine) est fabriqué par des cellules particulières, les mélanocytes, et distribué dans les cellules de la couche superficielle de la peau afin de « bloquer » la pénétration du rayonnement solaire pour nous protéger. Si un enfant est bronzé cela peut aussi diminuer la pénétration du rayonnement laser, donc atténuer l’efficacité du traitement. D’autre part certains mélanocytes ont pu malencontreusement être détruits (erreur de cible !) et la zone traitée peut rester blanche plusieurs mois. Après le laser, il est déconseillé de s’exposer au soleil pendant au moins un mois, car certains types de peaux (peaux mates) ont tendance à prendre une couleur marronne quand il y eu des phénomènes inflammatoires. C’est pour toutes ces raisons qu’on préfère faire du laser en hiver.
Est-ce que l’angiome peut refaire surface quelques années après l’arrêt des séances ?
De manière générale les angiomes plans ont tendance à s’épaissir avec le temps, notamment après la puberté, si on n’a pas pu détruire tous les vaisseaux avec le laser, l’angiome continue donc d’évoluer et on peut observer une recoloration. Il est donc souvent nécessaire de faire un entretien avec par exemple une séance tous les 2 ou 3ans.
Atteinte neurologique
Dans le syndrome de Sturge-Weber, certains enfants présentent une atteinte neurologique liée à une malformation des vaisseaux sanguins à la surface du cerveau, appelée angiome leptomeningée.
Cette anomalie peut perturber la circulation sanguine dans certaines zones du cerveau et entraîner différents symptômes neurologiques. Toutes les personnes atteintes par le syndrome Sturge-Weber ne présentent pas forcément cette atteinte.
Les manifestations neurologiques peuvent varier d’une personne à l’autre. Les plus fréquentes sont :
- Les crises d’épilepsie, qui apparaissent souvent dans la petite enfance
- Une faiblesse d’un côté du corps (appelée hémiparésie)
- Des migraines ou maux de tête
- Des difficultés d’apprentissage ou de concentration chez certains enfants
- Retard de croissance
La sévérité et la fréquence de ces symptômes peuvent être très différentes selon les patients. L’atteinte neurologique est généralement diagnostiquée grâce à des examens d’imagerie du cerveau, comme l’IRM.
Les enfants atteints du syndrome de Sturge-Weber sont suivis par un neurologue afin de surveiller l’évolution de la maladie et d’adapter les traitements si nécessaire.
Nous avons posé quelques questions au Docteur Sébastien Cabasson, Neuropédiatre de l’hôpital de Pau (Pyrénées-Atlantiques)
Quels sont les principaux signes neurologiques à surveiller chez un enfant atteint du syndrome de Sturge-Weber ?
Le premier risque du syndrome de Sturge-Weber est lié aux crises épileptiques, qui peuvent survenir précocement et vont surtout toucher le côté opposé à la lésion cutanée (les fibres nerveuses croisent la ligne médiane depuis le cortex vers la moelle épinière, à la partie basse du tronc cérébral). On parle dans ce cas de crises « focales », mais ces crises peuvent également être « généralisées », c’est-à-dire qu’elles vont provoquer une perte de conscience et des mouvements épileptiques des quatre membres.
Le second risque est l’apparition progressive d’un déficit de l’hémicorps opposé à l’angiome, avec une hémiparésie (faiblesse d’un côté) voire une hémiplégie dans les cas les plus sévères. Ces déficits apparaissent au départ à la suite des crises (on parle de « déficit post-critique ») mais peuvent survenir par la suite indépendamment des crises épileptiques et ressemblent à des accidents vasculaires cérébraux : on parle alors de « stroke-like » (stroke signifiant « attaque » en anglais, pour attaque cérébrale). Ces stroke-like sont au départ résolutif.
Quelle est la cause des crises d’épilepsie dans ce syndrome, et comment peuvent-elles évoluer avec l’âge ?
L’angiome de la pie-mère (la pie-mère est l’enveloppe des méninges la plus au contact du cortex cérébral, c’est elle comme son nom l’indique qui « nourrit » le cortex, provoque des anomalies micro-vasculaire, des zones mal irriguées, qui vont progressivement « souffrir », se calcifier, puis s’atrophier. Cela provoque donc des crises d’épilepsie qui sont le témoignage de cette « irritation » du cortex, le cortex comprenant les neurones qui sont à l’origine des crises épileptiques.
Que faire si on assiste à une crise d’épilepsie chez un adulte ou chez un enfant ?
Ne pas paniquer ! la plupart des crises épileptiques sont impressionnantes mais souvent brèves et sans conséquences pour l’oxygénation et le rythme cardiaque. Il faut essayer de mettre la personne en PLS (position latérale de sécurité) ou sur le dos, si possible à l’écart d’objets contondants. IL NE FAUT PAS METTRE D’OBJET DANS LA BOUCHE D’UN EPILEPTIQUE car on risque de casser des dents, se faire couper le doigt par la tétanie des mâchoires, voire entraîner l’inhalation de l’objet par le patient qui convulse. Si la personne était en train de manger on peut essayer d’enlever les aliments mais c’est souvent impossible tant que le patient est en crise : intérêt de la PLS. Un traitement d’urgence intra-rectal pour les nourrissons ou intra buccal pour les enfants plus grands peut être donné quand l’épilepsie est connue.
A quel moment faut-il envisager une imagerie cérébrale de contrôle (IRM, Scanner) et à quelle fréquence ?
L’IRM et le scanner (qui sont des examens complémentaires dans le syndrome de Sturge-Weber) sont réalisés très tôt lorsque le diagnostic est suspecté. La fréquence de contrôle dépend de l’évolution de chaque malade. Il peut y avoir une discordance entre les images et l’état clinique du patient. Il faut essayer de na pas faire trop de scanner (irradiation rayons X) à ces enfants.
Le syndrome a-t-il un impact sur le développement cognitif et moteur ? Comment peut-on l’anticiper ou le soutenir ?
Oui, le syndrome peut entraîner un retard intellectuel. Certains patients peuvent développer des syndromes autistiques. Néanmoins, chaque patient est différent, il est difficile de prévoir l’évolution, mais la prise en charge précoce, multidisciplinaire dans sa globalité, permettant d’améliorer le pronostic.
Existe-t-il des options chirurgicales pour les cas sévères d’épilepsie ou de malformation cérébrale ?
En effet certains patients avec des épilepsies très sévères et/ou une hémiplégie précoce peuvent bénéficier d’une prise en charge neurochirurgicale. Quelques équipes françaises proposent après un bilan rigoureux de « déconnecter » l’hémisphère atteint. Les convulsions ne sont plus visibles mais le patient gardera une faiblesse du côté opposé, qui pourra néanmoins s’améliorer avec la kinésithérapie mais également grâce aux quelques fibres neurologiques qui proviennent de l’autre hémisphère… et la plasticité neurologique des enfants.
Y a-t-il un risque accru d’accidents vasculaires cérébraux ou d’autres complications neurologiques ?
Les « pseudo-AVC » ou stroke-like sont fréquents dans le syndrome mais il ne s’agit pas à proprement parler d’AVC. L’autre symptomatologie neurologique fréquente est la migraine.
Atteinte oculaire (glaucome)
Le syndrome de Sturge-Weber peut être associé à un Glaucome, une maladie de l’œil liée à une augmentation de la pression à l’intérieur de l’œil.
Chez certains patients, les anomalies vasculaires du syndrome peuvent perturber l’écoulement du liquide de l’œil (humeur aqueuse). Cela peut entraîner une augmentation de la pression intraoculaire qui, si elle n’est pas traitée, peut endommager le nerf optique.
Le glaucome peut apparaître :
A la naissance ou dans la petite enfance
Plus tard dans l’enfance
Parfois à l’âge adulte
Un suivi ophtalmologique régulier est donc essentiel.
On estime qu’environ 30 à 70% des personnes atteintes du syndrome de Sturge-Weber peuvent développer un glaucome au cours de leur vie. Le risque le plus élevé lorsque l’angiome facial touche la zone de l’œil.
Le diagnostic est posé par un ophtalmologiste.
Les examens peuvent inclure :
Mesure de la pression intraoculaire
Examen du nerf optique
Mesure de la taille de l’œil
Examens sous anesthésie chez les jeunes enfants si nécessaire.
Nous avons pu poser quelques questions au Pr ……………….
- Peut-on guérir un glaucome ?
- Quelle est la cause du glaucome dans le cadre du syndrome Sturge-Weber ?
- Quels traitements médicaux sont le plus souvent utilisés chez les patients atteints de ce type de glaucome ?
- Quel est le pronostic visuel à long terme pour une personne atteint de glaucome associé au syndrome de Sturge-Weber ?
- Existe-t-il des signes à surveiller à la maison pour détecter une aggravation ou une augmentation de la pression oculaire ?
- Y a-t-il actuellement des essais cliniques ou des traitements innovants en développement pour ce type de glaucome ?
- Certains patients adultes doivent recourir à la pose d’un implant ou à d’autre dispositifs chirurgicaux. Cela peut-il être gênant au quotidien ?
- Les écrans, la lumière du soleil ou certaines activités peuvent-ils influencer la pression oculaire ? Y a-t-il des précautions particulières à prendre ?
- Quel pourriez-vous ajouter pour rassurer les patients ?
Le PNDS Syndrome Sturge-Weber
Le Protocole National de Diagnostic et de Soins (PNDS) consacré au syndrome de Sturge-Weber est un document de référence destiné aux professionnels de santé.
Il a été élaboré par des experts médicaux afin d’harmoniser la prise en charge des patients sur l’ensemble du territoire.
L’objectif du PNDS est d’aider les médecins et les équipes médicales à proposer une prise en charge adaptée et coordonnée aux personnes atteintes du syndrome de Sturge-Weber.
A la fin du PNDS figure une carte d’urgence patient.
Cette carte est destinée à être remplie par le médecin qui suit le patient et conservée par la personne concernée ou sa famille.
Elle permet, en cas d’urgence ou de consultation dans un autre service médical, de transmettre rapidement aux professionnels de santé les informations essentielles concernant la maladie, les traitements en cours et les précautions particulières à connaitre.
Il est indispensable de faire compléter cette carte par leur médecin et que les patients la gardent toujours sur eux.
Les centres coordinateurs
